Therapies Targeting ALS-Linked Genetic Mutations
The discovery of ALS-linked genetic mutations has opened the door to the development of genetically targeted therapies. These therapies are designed to reduce, block or eliminate the abnormal proteins produced from the altered instructions found in these genes. As researchers learn more about the DNA changes and toxic proteins that drive the development of ALS, they will be better able to target therapies to treat the underlying causes of the disease.
Mutated Genes Create Toxic Proteins
An estimated 60% of people with familial ALS and about 10% of people with sporadic ALS have a mutation in one of the more than 40 genes linked to the disease. Some of these genes include SOD1, C9orf72, FUS, TARDBP and ATXN2.
A mutation changes the letters in the DNA (also known as the genetic sequence) that spell out the instructions for making a specific protein. When a cell needs to make this protein, these genetic “typos” are copied from the DNA into shorter related molecules called RNA during a process called transcription. The RNA then serves as a messenger, taking the instructions from the nucleus to structures called ribosomes, which serve as the protein-making factories of cells. In the ribosome, this information is translated from the language of genetic code into the language of amino acids, which are the building blocks of proteins. If this information includes any mistakes (mutations), it changes the protein that is produced.
In the case of SOD1, for example, the gene contains the instructions for producing a protein called superoxide dismutase type 1 (SOD1). This protein normally helps clear out toxins from the brain, but when the SOD1 gene is mutated, it creates a toxic SOD1 protein. The abnormal protein not only fails to do its normal job, but it also creates its own problems. In people with SOD1 mutations, the defective SOD1 proteins that are produced clump together, and these clumps, known as aggregates, damage the nervous system, leading to the development of ALS.
How Gene-Targeted Therapies Work
To stop the damage caused by mutations in ALS-linked genes, researchers are leveraging a number of different technologies that reduce, block or eliminate toxic proteins and their aggregates. They do this by specifically targeting mutated genes (DNA), messenger RNA (mRNA) or the toxic protein itself.
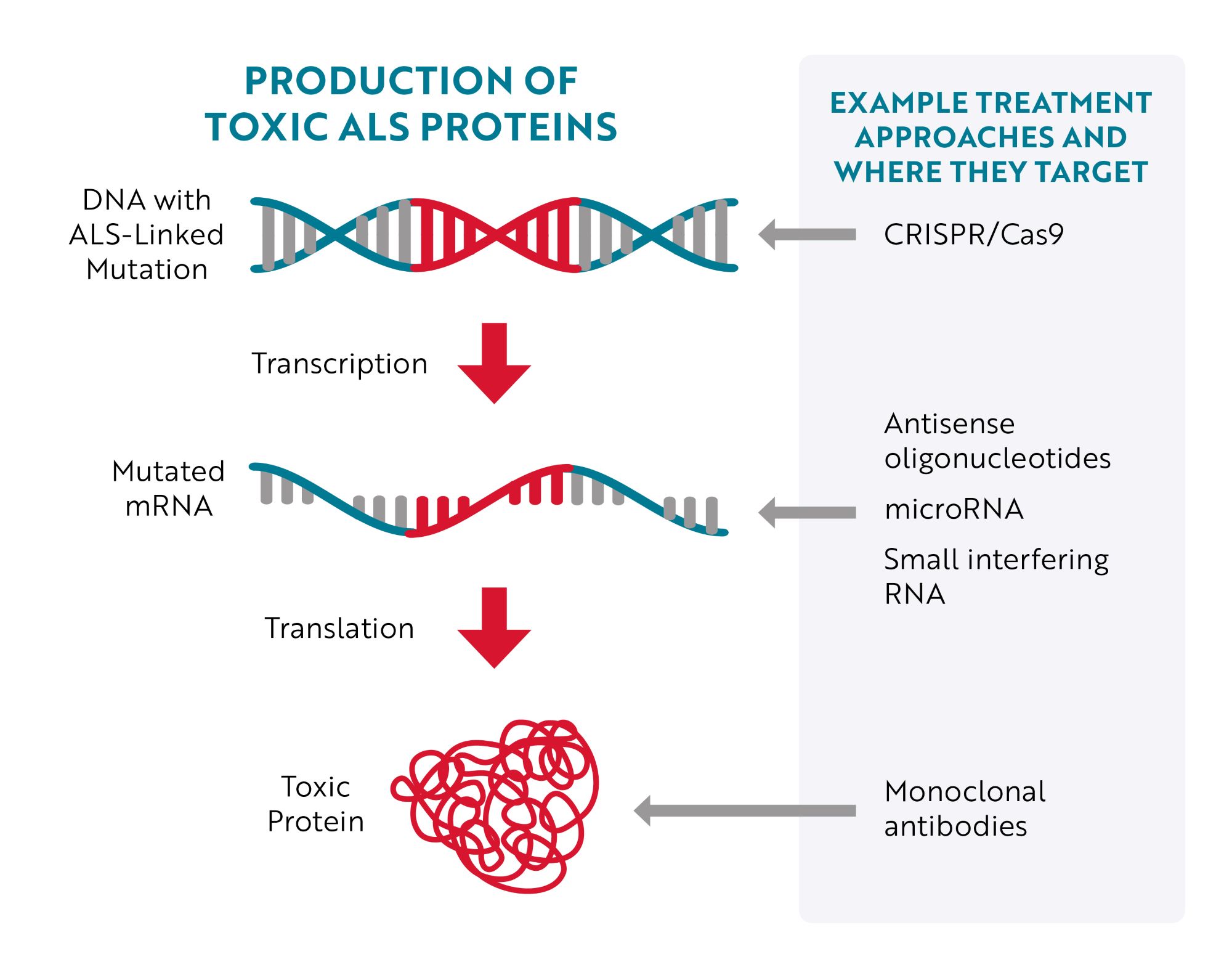
Targeting DNA
The most obvious place to target would be the source of the issue – the mutated gene (DNA) – but until recently, that wasn’t easily accomplished. With the development of CRISPR/Cas9 gene editing technology, researchers have more efficient tools to target and correct specific mutations. Essentially, researchers can program CRISPR/Cas9 gene editing tools to “cut out” a specific region of DNA. For example, in a preclinical, “proof-of-concept" study in mouse models of ALS and human cells, researchers were able to remove the repeat expansion in the C9ORF72 gene, the most common genetic cause of ALS, without affecting the parts of the gene that provide instructions for making healthy C9ORF72 proteins.
Targeting RNA
An approach much further along in development is to target mutated messenger RNA (mRNA). The majority of therapies being tested to treat ALS caused by a genetic mutation specifically bind to mutated mRNA, telling cells that the mRNA needs to be destroyed before it can be turned into toxic proteins.
This type of therapy is called gene silencing because, instead of repairing or interacting with the mutated gene (DNA), it aims to “silence” the gene’s effect. Antisense oligonucleotides (ASOs), microRNA (miRNA) and short interfering RNA (siRNA) are all different therapeutic approaches designed to achieve this goal.
Despite the promise of being able to stop the production of toxic proteins, these therapies will probably not be able to simply turn off ALS like a light switch. Research is showing that it takes time for these therapies to reduce protein levels and even more time after that for this reduction to be translated into a clinically meaningful benefit, such as slowing disease progression, improving physical function or living longer.
Targeting Toxic Proteins
A third approach is to target the toxic protein after it has been produced. An example of this approach is the investigational treatment AP-101. AP-101 is a human monoclonal antibody. Monoclonal antibodies are proteins made in laboratories that act like the antibodies produced by the immune system. Antibodies made by the immune system seek out a wide range of foreign and toxic substances, like bacteria and viruses, and then stick to them so the body can destroy them. Laboratory-made monoclonal antibodies are created to seek out and destroy one specific thing. In the case of AP-101, it was designed to selectively target and reduce toxic clumps of misfolded SOD1 proteins.
In early laboratory studies, AP-101 reduced motor symptoms and increased survival in a mouse model of ALS. A subsequent phase 1 study showed it was well-tolerated by people with ALS at all tested doses. AP-101 is now being evaluated in a phase 2 study involving 63 participants with both familial and sporadic ALS with results expected in the second half of 2023.
Opportunities for Sporadic ALS
These approaches are not limited to people with ALS-linked gene mutations. Researchers are interested in finding ways to prevent or dissolve toxic clumps of other ALS-related proteins, such as TDP-43, which are found in 97% of all people with ALS. Learn more about this research.
Focusing on Prevention
Because toxic proteins build up and cause damage over time, researchers are interested in finding out whether targeted therapies could benefit people with ALS-linked gene mutations but no signs or symptoms of ALS. The thought is that by treating people with these mutations before significant and irreversible neuron damage occurs, it could delay or even prevent the onset of ALS.
The first clinical trial for pre-symptomatic ALS, the phase 3 ATLAS Study, is testing tofersen in approximately 150 participants who have specific SOD1 gene mutations but do not have any signs or symptoms of ALS. Through this study, the researchers are trying to determine whether tofersen can delay the onset of signs or symptoms of ALS and/or slow declines in function once signs or symptoms appear. The trial is projected to be completed in 2027.
Therapies in Clinical Trials
Last updated March 2024
| Drug Name | Therapy Type | Target | Phase | Estimated Completion |
|---|---|---|---|---|
| BIIB067 (tofersen) | ASO | SOD1 mRNA | Phase 3 (pre-symptomatic gene carriers) | 2027 |
| ION-363 (jacifusen) | ASO | FUS mRNA | Phase 3 | 2028 |
| AP-101 | Monoclonal antibody | Misfolded and aggregated SOD1 protein | Phase 2* | 2025 |
| BIIB105 / ION-541 | ASO |
ATXN2 mRNA |
Phase 1/2* | 2026 |
| AMT-162 | miRNA | SOD1 mRNA | Phase 1/2 | 2032 |
* Also being tested for people with sporadic ALS
Importance of Genetic Testing
Because of the targeted nature of these therapies, they only work for people who have mutations in the specific gene the therapy was developed for. The only way to know if you have a mutation in an ALS-linked gene is to get a genetic test.
The decision whether to get tested or not is a very personal one. Genetic testing comes with benefits, but also with risks, and may not be right for you. A genetic counselor can help you weigh the pros and cons as you decide whether or not to get tested. Click here to learn more about the benefits and risks of genetic testing for people currently living with ALS. If you haven’t been diagnosed with ALS but have family members with the disease, click here to explore the potential benefits and risks of genetic testing.
ALS Association Support
We have made significant investments over the years into identifying the underlying genetic causes of ALS. This support led to the landmark discoveries of the SOD1 gene mutations in 1993 and C9orf72 in 2011, the most common gene associated with ALS. Since then, multiple large, global “big data” initiatives we’ve supported, such as the New York Genome Center and Project MinE, have undertaken large sequencing and gene identification efforts. This work has led to the discovery of additional genes thought to cause or increase the risk of developing ALS, and therefore increasing the number of potential targets for genetically targeted therapies.
Thanks to the generosity of our donors, we also have been able to fund numerous research groups that are exploring the use of antisense and other DNA and RNA-based therapies to target a diverse array of ALS-linked genes and proteins. We continue to be excited by the potential of these therapies to improve the lives of people with both familial and sporadic ALS as well as other neurodegenerative diseases.
