Antisense Therapy for ALS
Antisense therapy is a way to treat ALS and other neurologic conditions using short DNA-like molecules called antisense oligonucleotides (ASOs). ASOs have emerged as a promising treatment approach following the FDA’s approval of Spinraza for spinal muscular atrophy (SMA) in 2016. SMA is a common neuromuscular disease that is the leading cause of genetic death in infants and toddlers. The success of Spinraza provides hope for the development and approval of effective antisense therapies for people living with ALS.
Making Sense of Antisense
ASOs are short strings of nucleotides – the chemical building blocks of our genetic code – that are designed to interact with RNA. RNA acts as a messenger, taking the instructions found in genes (DNA) to the cell’s protein-making factories (ribosomes). Because the instructions spelled out by the nucleotides of messenger RNA (mRNA) can be translated into a protein, it is known as a “sense” sequence. ASOs, on the other hand, get their name because scientists build them in a complementary “antisense” manner.

Because of their complementary design, ASOs seek out and attach to a specific sequence of mRNA, allowing researchers to precisely target the therapy. Once an ASO binds to mRNA, it can do one of two things: “knock down” the mRNA or modulate its splicing.
Knockdown ASOs
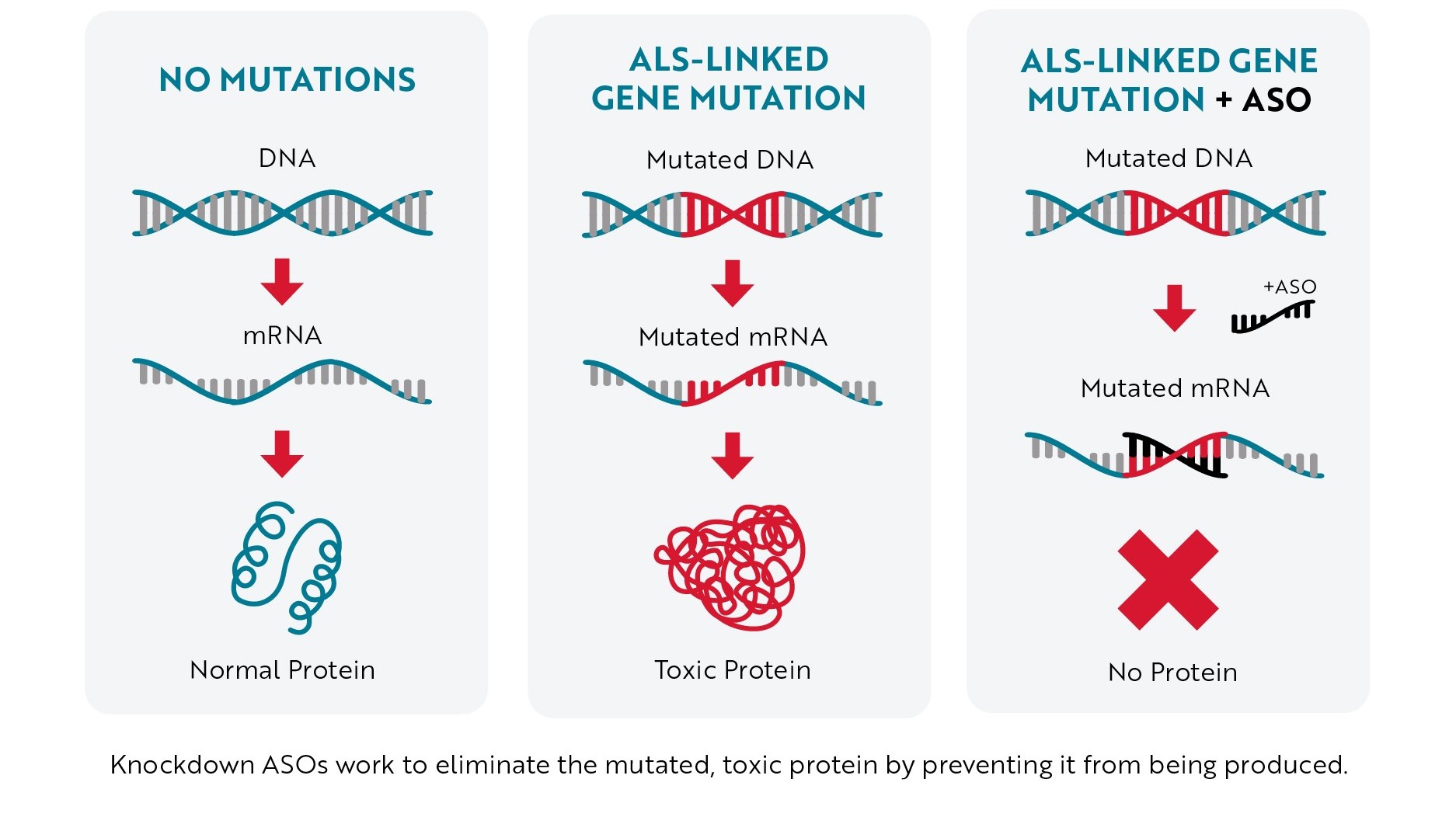
Most of the ASOs in development to treat ALS are what scientists call “knockdown ASOs.” When this type of ASO binds to mRNA (usually mRNA created from a mutated gene), it marks the mRNA for destruction before it can be turned into a toxic protein. Without the toxic protein being produced, neurons have a better chance of staying healthy, helping slow or even stop ALS progression.
Antisense Technology – Knockdown Approach
Splice-Modulating ASOs
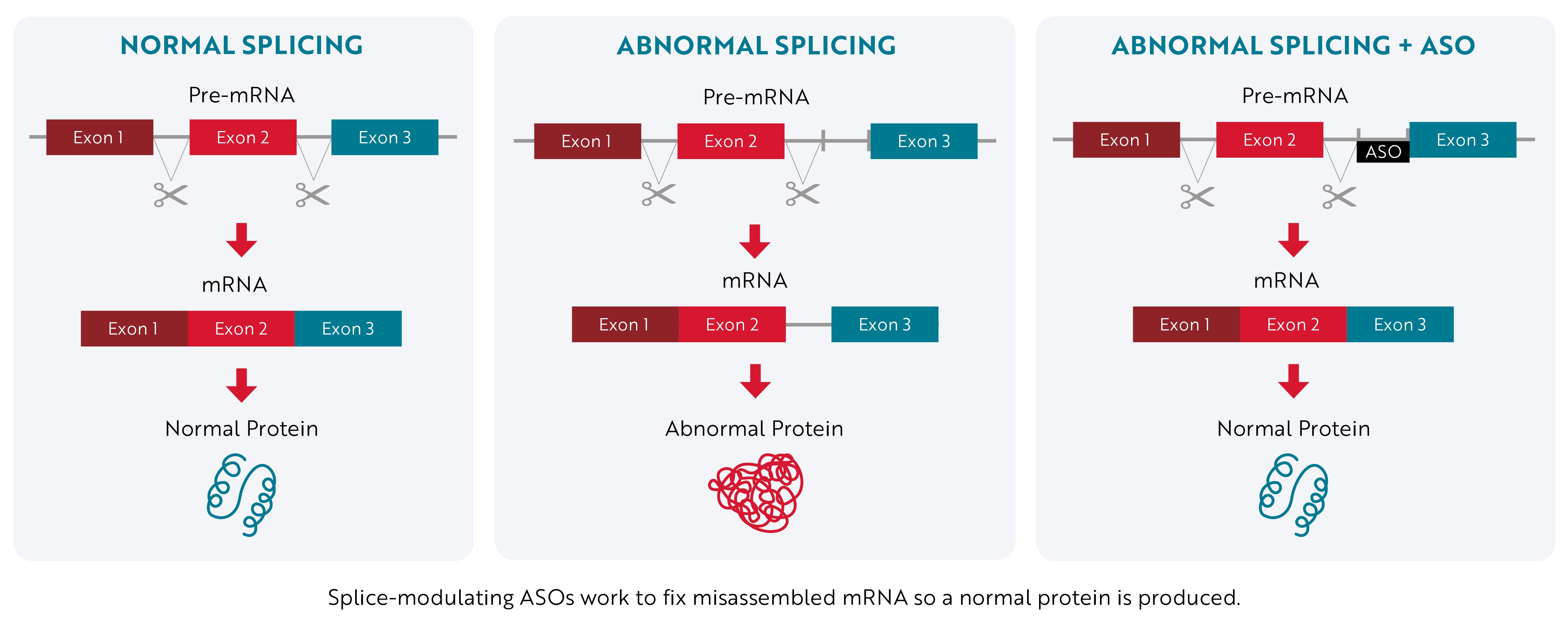
Instead of stopping a toxic protein from being made, splice-modulating ASOs attempt to promote the production of a normal protein by correcting a mistake that occurs during mRNA assembly.
When a gene’s instructions are copied from DNA into RNA, they contain essential information called exons and nonessential information called introns. The “first draft” of RNA that’s assembled, known as pre-mRNA, contains both exons and introns. Then, during a process called RNA splicing, the cell edits the instructions by removing the nonessential introns. Once the introns are removed, the exons are joined together to form the final instructions (mRNA) that are used to make proteins.
Sometimes, the cell makes mistakes during this editing process, which affects the protein that is produced. Splice-modulating ASOs work by binding to the mRNA and helping the cell cut out all the introns correctly so that a normal protein is produced.
Antisense Technology – Splice-Modulating Approach
ASOs and Familial ALS
The discovery and sequencing of more than 40 different genes linked to ALS has provided researchers with an initial list of possible targets for antisense therapy. So far, ASOs targeting four of these genes are in various stages of development: C9orf72, the most common genetic cause of ALS; SOD1, the second-most common cause of familial ALS; FUS and ATXN2.
Learn more about therapies targeting ALS-linked genetic mutations.
ASOs and Sporadic ALS
By studying familial ALS, researchers have uncovered the important role RNA production and processing play in ALS development and progression. With this knowledge has come hope that ASOs could be used to treat sporadic ALS as well. Much of this research centers on an RNA-binding protein called TDP-43.
TDP-43 is a tantalizing target for researchers because abnormal clumps of this protein (also known as aggregates) are found in nearly all (97%) people living with ALS. Usually, TDP-43 helps keep cells in the central nervous system healthy by regulating the protein production process from within the nucleus (central structure that contains a cell’s DNA). In ALS, TDP-43 exits the nucleus, becomes misshapen and clumps into aggregates in the surrounding cytoplasm. This not only means that TDP-43 can’t perform its usual healthy function, but the aggregates also block other normal cellular processes and become toxic to the cell, leading to neurodegeneration.
Preventing Aggregation
Since TDP-43 plays a vital role in keeping neurons healthy, researchers aren’t able to block cells from producing this protein as a way to treat ALS. However, preclinical research has shown that using knockdown ASOs to stop the production of another related protein called ataxin-2 can significantly decrease the number of TDP-43 aggregates in neurons.
BIIB105 / ION541 is an investigational ASO designed to reduce ataxin-2 levels in motor neurons. It binds to mRNA produced from the ATXN2 gene, telling neurons that the mRNA needs to be destroyed before it can be turned into a protein. In mouse models of ALS, treatment with BIIB105 / ION541 helped these animals live longer. BIIB105 / ION541 is currently being tested in a phase 1/2 clinical trial, known as the ALSpire study, by participants who have a mutation in the ATXN2 gene as well as participants with sporadic ALS. The study is expected to end in 2026.
Fixing mRNA Mistakes
One of the important roles TDP-43 plays in neurons is directing the RNA splicing process. When TDP-43 disappears from the nucleus, pieces of information that would normally have been edited out of pre-mRNA are included (see above). So, while the gene itself is completely normal (no mutations), mistakes are introduced into the mRNA sequence in the absence of TDP-43. These errors often produce shorter, non-working versions of important proteins like stathmin-2 and UNC13A that can drive ALS development and progression.
Based on this knowledge, researchers are developing splice-modulating ASOs they hope will correct these mistakes in mRNA, restore normal protein levels, and help slow or stop neurodegeneration.
One example is QRL-201, which aims to restore stathmin-2 levels and function in people with ALS. Researchers have found that almost all people with ALS have lower-than-normal levels of this protein. Stathmin-2 is essential for maintaining the connection between motor neurons and muscle. Without stathmin-2, motor neurons disconnect from muscle, causing the paralysis that is characteristic of ALS. Preclinical studies have shown that QRL-201 helps increase stathmin-2 to normal levels and reverse a number of neurodegenerative processes in lab-grown motor neurons affected by TDP-43 aggregation. A first-in-human, phase 1 study of QRL-201 based in Canada is expected to be completed in May 2025.
Other research groups and biotechnology companies are also developing ASOs directed at stathmin-2 and UNC13A.
ALS Association Support
We were the first to fund research of ALS-specific ASOs back in 2004 when it was an emerging technology, despite the high risk of the technology not coming to fruition. We subsequently committed more than $1.3 million to ASO technology and funded five additional studies, including preclinical, safety and pharmacodynamic measurements and the first-in-human phase 1 trial assessing safety and target engagement for an early SOD1-targeting antisense drug, ISIS 333611. These studies supported continued investigation of SOD1 as a therapeutic target and led to the development of the more potent antisense drug, tofersen, now known on Qalsody.
We also partnered with Project ALS to support a pilot clinical research project of ION363 (jacifusen), an experimental therapy for ALS caused by a mutation in the FUS gene. This funding made it possible for Dr. Neil Shneider and his team at Columbia University’s Eleanor and Lou Gehrig ALS Center to offer the therapy to 10 people with FUS-associated ALS over two years while tracking safety data on the drug and disease-relevant biomarkers. This research led to Ionis Pharmaceuticals launching a pivotal phase 3 clinical trial of ION363 in 2021.
Thanks to the generosity of our donors, we have been able to fund a number of other research groups exploring the use of ASOs to target a diverse array of genes and proteins, including projects focused on treating sporadic ALS. We continue to be excited by the potential of antisense therapy to improve the lives of people with ALS and other neurodegenerative diseases. We are hopeful ASOs will serve as a valuable tool in making ALS a livable disease for everyone, everywhere.

